
In brief
MIT chemical engineers have found a way to speed up a chemical reaction that is key to converting captured carbon dioxide emissions into useful, valuable products. To prepare, they add a bit of DNA to a commonly used catalyst. They then rely on the natural behavior of DNA to situate the catalyst where it’s needed on an electrode inside an electrochemical device. The DNA-linked catalyst plus a jolt of voltage provides the encouragement needed to make the reluctant reaction happen. The team found that the modified catalyst is more stable under typical operating conditions, and it significantly improves the conversion efficiency. The researchers believe that their approach can be used with other classes of catalysts to encourage a wide range of reluctant electrochemical reactions.
Carbon dioxide (CO2) is a major contributor to climate change and a significant product of many human activities, notably industrial manufacturing. A major goal in the energy field has been to chemically convert emitted CO2 into valuable chemicals or fuels. But while CO2 is available in abundance, it has not yet been widely used to generate value-added products. Why not?
The reason is that CO2 molecules are highly stable and therefore not prone to being chemically converted to a different form. Researchers have sought materials and device designs that could help spur that conversion, but nothing has worked well enough to yield an efficient, cost-effective system.
Two years ago, Ariel Furst, the Raymond (1921) and Helen St. Laurent Career Development Professor of Chemical Engineering at MIT, decided to try using something different—a material that gets more attention in discussions of biology than of chemical engineering. Already, results from work in her lab suggest that her unusual approach is paying off.
The stumbling block
The challenge begins with the first step in the CO2 conversion process. Before being transformed into a useful product, CO2 must be chemically converted into carbon monoxide (CO). That conversion can be encouraged using electrochemistry, a process in which input voltage provides the extra energy needed to make the stable CO2 molecules react. The problem is that achieving the CO2-to-CO conversion requires large energy inputs—and even then, CO makes up only a small fraction of the products that are formed.
To explore opportunities for improving this process, Furst and her research group focused on the electrocatalyst, a material that enhances the rate of a chemical reaction without being consumed in the process. The catalyst is key to successful operation. Inside an electrochemical device, the catalyst is often suspended in an aqueous (water-based) solution. When an electric potential (essentially a voltage) is applied to a submerged electrode, dissolved CO2 will—helped by the catalyst—be converted to CO.
But there’s one stumbling block: The catalyst and the CO2 must meet on the surface of the electrode for the reaction to occur. In some studies, the catalyst is dispersed in the solution, but that approach requires more catalyst and isn’t very efficient, according to Furst. “You have to both wait for the diffusion of CO2 to the catalyst and for the catalyst to reach the electrode before the reaction can occur,” she explains. As a result, researchers worldwide have been exploring different methods of “immobilizing” the catalyst on the electrode.
Connecting the catalyst and the electrode
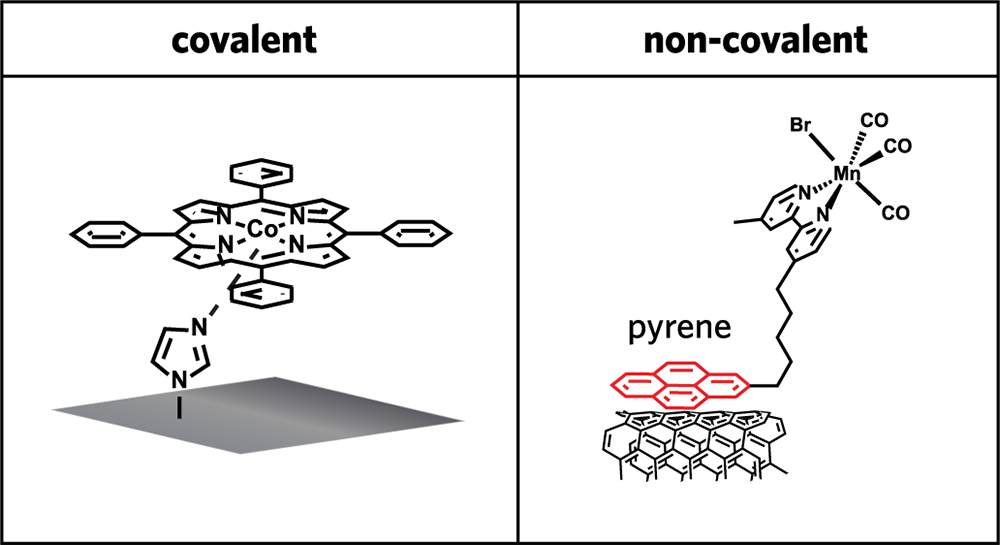
Two common approaches to immobilizing small-molecule catalysts on electrodes. In the left-hand drawing, the catalyst at the top is connected to a glassy carbon electrode by a nitrogen-based ring; the two structures share electrons, forming a covalent bond that is essentially permanent. In the right-hand drawing, the catalyst is connected to an electrode of carbon nanotubes by a pyrene molecule; because the pyrene and carbon nanotubes don’t share electrons, they are attached non-covalently and can be separated easily.
Before Furst could delve into that challenge, she needed to decide which of the two types of CO2 conversion catalysts to work with: the traditional solid-state catalyst or a catalyst made up of small molecules. In examining the literature, she concluded that small-molecule catalysts held the most promise. While their conversion efficiency tends to be lower than that of solid-state versions, molecular catalysts offer one important advantage: They can be tuned to emphasize reactions and products of interest.
The figure above illustrates two common approaches to immobilizing small-molecule catalysts on an electrode. In the left-hand drawing, the metal center of the catalyst at the top (here, cobalt) is linked by a nitrogen-based ring to a glassy carbon electrode (the gray square) by strong covalent bonds—a type of bond in which atoms share electrons; the result is a strong, essentially permanent connection. In the right-hand drawing, a molecule (in this case, pyrene) sets up a non-covalent attachment between the manganese-based catalyst at the top and an electrode made of carbon materials (in this case, nanotubes); unlike a covalent bond, this connection can easily be broken.
Neither approach is ideal. In the former case, the catalyst and electrode are firmly attached, ensuring efficient reactions; but when the activity of the catalyst degrades over time (which it will), the electrode can no longer be accessed. In the latter case, a degraded catalyst can be removed; but the exact placement of the small molecules of the catalyst on the electrode can’t be controlled, leading to an inconsistent, often decreasing, catalytic efficiency—and simply increasing the amount of catalyst on the electrode surface without concern for where the molecules are placed doesn’t solve the problem.
What was needed was a way to position the small-molecule catalyst firmly and accurately on the electrode and then release it when it degrades. For that task, Furst turned to what she and her team regard as a kind of “programmable molecular Velcro”: deoxyribonucleic acid, or DNA.
Adding DNA to the mix
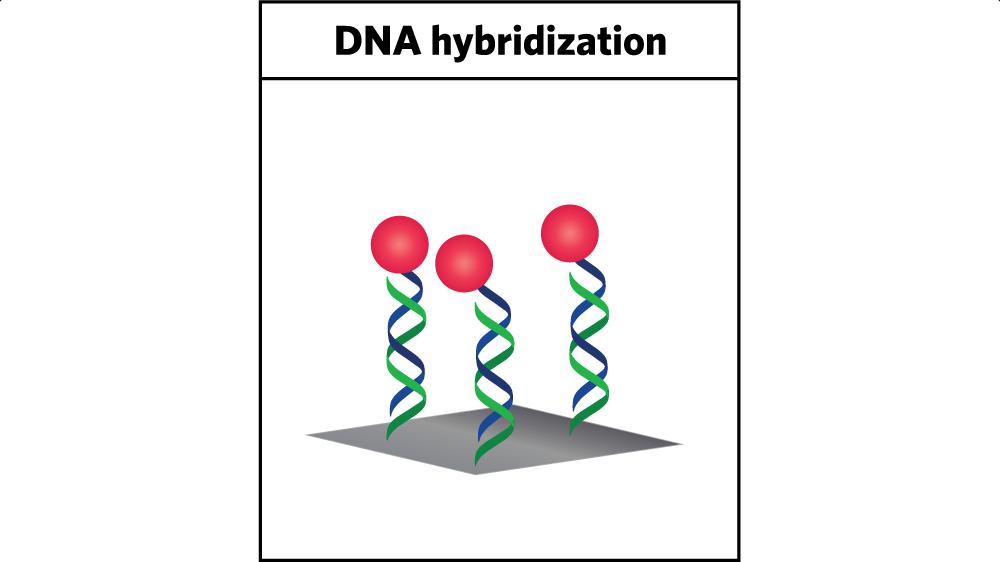
Using DNA to attach the catalyst to the electrode. In the researchers’ approach, the catalyst—represented above by the red sphere—is connected to a carbon electrode by strands of DNA. Initially, one strand of DNA is deposited on the electrode, and the other is in solution with the catalyst attached. When the two strands meet up, they lock together, as shown here. The green strand that is attached to the electrode and the blue strand that is carrying the catalyst are now connected, so the catalyst is firmly linked to the electrode. Over time, the catalyst will become inactive. Raising the temperature will release the DNA strand with the spent catalyst so fresh catalyst can be attached.
Mention DNA to most people, and they think of biological functions in living things. But the members of Furst’s lab view DNA as more than just genetic code. “DNA has these really cool physical properties as a biomaterial that people don’t often think about,” she says. “DNA can be used as a molecular Velcro that can stick things together with very high precision.”
Furst knew that DNA sequences had previously been used to immobilize molecules on surfaces for other purposes. So she devised a plan to use DNA to direct the immobilization of catalysts for CO2 conversion.
Her approach depends on a well-understood behavior of DNA called hybridization. The familiar DNA structure is a double helix that forms when two complementary strands connect. When the sequence of bases (the four building blocks of DNA) in the individual strands match up, hydrogen bonds form between complementary bases, firmly linking the strands together.
Using that behavior for catalyst immobilization involves two steps. First, the researchers attach a single strand of DNA to the electrode. Then they attach a complementary strand to the catalyst that is floating in the aqueous solution. When the latter strand gets near the former, the two strands hybridize; they become linked by multiple hydrogen bonds between properly paired bases. The result is shown in the figure above. The catalyst (shown as a red sphere) is firmly affixed to the electrode by means of two interlocked, self-assembled DNA strands (the wavy lines), one connected to the electrode (shown in green) and the other to the catalyst (shown in blue).
Better still, the two strands can be detached from one another. “The connection is stable, but if we heat it up, we can remove the secondary strand that has the catalyst on it,” says Furst. “So we can de-hybridize it. That allows us to recycle our electrode surfaces—without having to disassemble the device or do any harsh chemical steps.”
Experimental investigation
To explore that idea, Furst and her team—postdoctoral researchers Gang Fan and Thomas Gill, former graduate student Nathan Corbin PhD ’21, and former postdoc Amruta Karbelkar—performed a series of experiments using three small-molecule catalysts based on porphyrins, a group of compounds that are biologically important for processes ranging from enzyme activity to oxygen transport. Two of the catalysts involve a synthetic porphyrin plus a metal center of either cobalt or iron. The third catalyst is hemin, a natural porphyrin compound used to treat porphyria, a set of disorders that can affect the nervous system. “So even the small-molecule catalysts we chose are kind of inspired by nature,” comments Furst.
In their experiments, the researchers first needed to modify single strands of DNA and deposit them on one of the electrodes submerged in the solution inside their electrochemical cell. Though this sounds straightforward, it did require some new chemistry. Led by Karbelkar and undergraduate researcher Rachel Ahlmark ’24, the team developed a fast, easy way to attach DNA to electrodes. For this work, the researchers’ focus was on attaching DNA, but the “tethering” chemistry they developed can also be used to attach enzymes (protein catalysts), and Furst believes it will be highly useful as a general strategy for modifying carbon electrodes.
Once the single strands of DNA were deposited on the electrode, the researchers synthesized complementary strands and attached to them one of the three catalysts. When the DNA strands with the catalyst were added to the solution in the electrochemical cell, they readily hybridized with the DNA strands on the electrode. After half an hour, the researchers applied a voltage to the electrode to chemically convert CO2 dissolved in the solution and used a gas chromatograph to analyze the makeup of the gases produced by the conversion.
The team found that when the DNA-linked catalysts were freely dispersed in the solution, they were highly soluble—even when they included small-molecule catalysts that don’t dissolve in water on their own. Indeed, while porphyrin-based catalysts in solution often stick together, once the DNA strands were attached, that counterproductive behavior was no longer evident.

Improving catalyst stability by adding single DNA strands to a catalyst in solution. The stability of a catalyst can be tested by inputting a voltage onto an electrode inside an electrochemical device and monitoring the resulting current that flows. Left panel: When a voltage of practical interest was applied to a test device containing unmodified catalyst in solution, the measured current rapidly dropped over time, indicating that the free-floating catalyst was degrading—a major problem in an electrochemical system. Right panel: When the floating catalyst had DNA strands attached, that rapid decline in the measured current did not occur.
The DNA-linked catalysts in solution were also more stable than their unmodified counterparts. They didn’t degrade at voltages that caused the unmodified catalysts to degrade. “So just attaching that single strand of DNA to the catalyst in solution makes those catalysts more stable,” says Furst. “We don’t even have to put them on the electrode surface to see improved stability.” When converting CO2 in this way, a stable catalyst will give a steady current over time. Experimental results presented in the figure above show that adding the DNA prevented the catalyst from degrading at voltages of interest for practical devices. Moreover, with all three catalysts in solution, the DNA modification significantly increased the production of CO per minute.
Allowing the DNA-linked catalyst to hybridize with the DNA connected to the electrode brought further improvements, even compared to the same DNA-linked catalyst in solution. For example, as a result of the DNA-directed assembly, the catalyst ended up firmly attached to the electrode, and the catalyst stability was further enhanced. Despite being highly soluble in aqueous solutions, the DNA-linked catalyst molecules remained hybridized at the surface of the electrode, even under harsh experimental conditions.
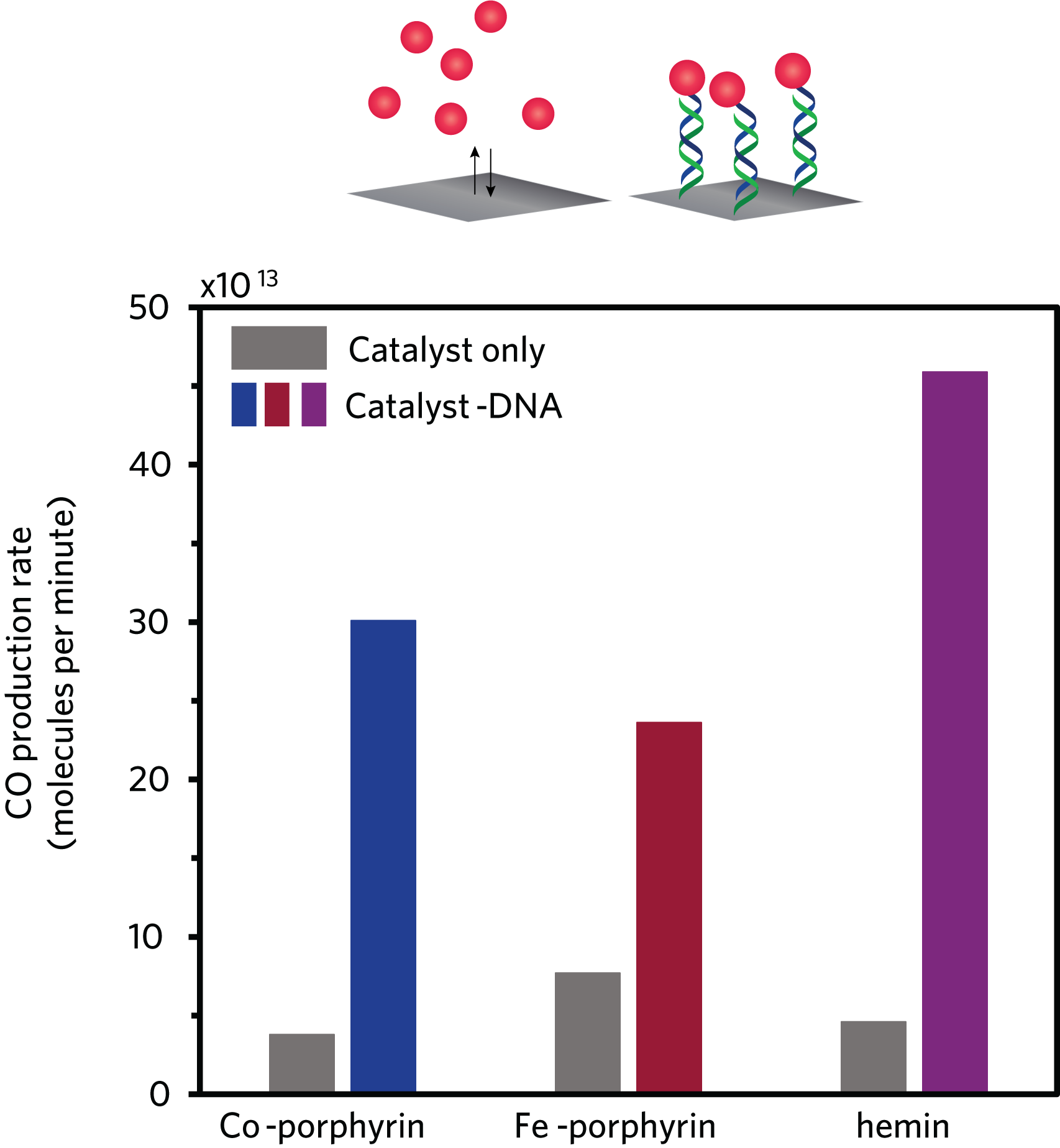
Impact on CO generation of immobilizing catalysts with DNA. In this figure, each pair of bars presents results with one of the three catalysts tested. The left-hand bar shows CO generated per minute with the unmodified catalyst in solution. The right-hand bar shows CO generated per minute with the same catalyst modified with DNA and immobilized on the electrode. Immobilization significantly increases the rate of CO output, regardless of the molecule used as the catalyst.
Immobilizing the DNA-linked catalyst on the electrode also significantly increased the rate of CO production. The figure above shows the CO production rate with each of the team’s catalysts in solution without attached DNA strands—the conventional setup—and then with them immobilized by DNA on the electrode. With all three catalysts, the amount of CO generated per minute was far higher when the DNA-linked catalyst was immobilized on the electrode.
In addition, immobilizing the DNA-linked catalyst on the electrode greatly increased the “selectivity” in terms of the products. One persistent challenge in using CO2 to generate CO in aqueous solutions is that there is an inevitable competition between the formation of CO and the formation of hydrogen. That tendency was eased by adding DNA to the catalyst in solution—and even more so when the catalyst was immobilized on the electrode using DNA. For both the cobalt-porphyrin catalyst and the hemin-based catalyst, the formation of CO relative to hydrogen was significantly higher with the DNA-linked catalyst on the electrode than in solution. With the iron-porphyrin catalyst they were about the same. “With the iron, it doesn’t matter whether it’s in solution or on the electrode,” Furst explains. “Both of them have selectivity for CO, so that’s good, too.”
Progress and plans

The researchers test and compare the performance of their catalysts using this sealed electrochemical cell. Here, an electrode modified with the catalyst is readied for testing. Once the electrochemical reaction begins, the generated gases will collect in the space above the solution, where they can be sampled and analyzed. Credit: Gretchen Ertl
Furst and her team have now demonstrated that their DNA-based approach combines the advantages of the traditional solid-state catalysts and the newer small-molecule ones. In their experiments, they achieved the highly efficient chemical conversion of CO2 to CO and also were able to control the mix of products formed. And they believe that their technique should prove scalable: DNA is inexpensive and widely available, and the amount of catalyst required is several orders of magnitude lower when it’s immobilized using DNA.
Based on her work thus far, Furst hypothesizes that the structure and spacing of the small molecules on the electrode may directly impact both catalytic efficiency and product selectivity. Using DNA to control the precise positioning of her small-molecule catalysts, she plans to evaluate those impacts and then extrapolate design parameters that can be applied to other classes of energy-conversion catalysts. Ultimately, she hopes to develop a predictive algorithm that researchers can use as they design electrocatalytic systems for a wide variety of applications.
This research was supported by a grant from the MIT Energy Initiative Seed Fund. Further information can be found in:
G. Fan, N. Corbin, T. Gill, A. Karbelkar, and A. Furst. “DNA-based immobilization for improved electrochemical carbon dioxide reduction.” ChemRxiv, Cambridge Open Engage, 2022. Online: doi.org/10.26434/chemrxiv-2022-qll2k.
This article appears in the Spring 2022 issue of Energy Futures.