
MIT and Leiden University researchers have now produced unambiguous experimental evidence that conventional theory doesn’t accurately describe how highly efficient metal-oxide catalysts help release oxygen gas from water during electrolysis—a critical process in many energy storage technologies. Using a special form of oxygen as a marker, they demonstrated that the oxygen gas comes not only from the water but also from the metal-oxide catalyst itself. In parallel theoretical studies, they showed that certain electronic properties of such highly efficient catalysts permit involvement of oxygen from the catalyst. The team is now working to define the sequence of chemical reactions that leads to oxygen release on these special catalysts. Already, their findings provide new guidance in the ongoing search for low-cost, effective materials and designs for these important catalysts.
Many systems for storing energy rely on electrochemical reactions that cause the release of oxygen gas from water. These so-called oxygen-evolution reactions are critical to the efficiency of devices that split water to recover hydrogen fuel and to the performance of regenerative fuel cells, metal-air batteries, and more.
“Oxygen evolution is one universal reaction that’s key in developing efficient energy storage technologies by storing electron energy in chemical form,” says Yang Shao-Horn, the W.M. Keck Professor of Energy, a professor of mechanical engineering and of materials science and engineering, and co-director of the MIT Energy Initiative’s Energy Storage Research Center.
The kinetics of oxygen-evolution reactions are typically slow, so catalysts such as metal oxides are required to speed up the chemical reactions (without being consumed in the process). During electrolysis of water, a metal-oxide catalyst is immersed in a water-based electrolyte. When a potential is applied, the water molecules react on the catalyst, splitting into positively charged hydrogen ions (protons) and oxygen atoms, which form oxygen gas that bubbles out of the system.
Much research has focused on identifying metal oxides that will perform this task. Many compounds work, but the catalytic activity of some is orders of magnitude greater than that of others. After three years of intensive experimental and theoretical study, Shao-Horn and her colleagues in the Electrochemical Energy Lab have developed new insights into why certain metal-oxide catalysts work so well, and they have produced practical guidelines for finding new, more effective catalysts in the future.

This electrochemical cell is at the core of the experimental setup shown in the first photo in this article. At its center, a probe containing the sample catalyst is submerged in the water-based electrolyte. A voltage is applied via the incoming wires, electrolysis commences, bubbles of oxygen gas form on the surface of the catalyst, and the resulting current flow is recorded. The probe spins rapidly to prevent bubbles from accumulating on the catalyst, which would stop the electrochemical reaction. Photo: Stuart Darsch
Experimental evidence: Tracking the oxygen
In their work, Shao-Horn, Binghong Han PhD ’16, former postdoc Alexis Grimaud, Visiting Professor Livia Giordano from Milano-Bicocca University in Italy, and their collaborators have been exploring a promising class of catalytic materials known as perovskites, which—unlike today’s state-of-the-art catalysts—consist of low-cost, earth-abundant materials. To understand what determines catalyst efficiency, they wanted to compare the behavior of two perovskites, one more catalytically active than the other. So they tested catalysts based on the same transition metal—cobalt—in combination with lanthanum and with strontium. The combination of cobalt, strontium, and oxygen is known to exhibit especially high catalytic activity—far higher than the lanthanum mixture.
A first question concerns the source of the oxygen that becomes oxygen gas. The conventional theory of metal-oxide catalysis says that all of the oxygen for the oxygen-evolution reactions comes from the water and that all of those reactions occur on the transition metal atoms—not the oxygen atoms—at the surface of the catalyst. But with certain materials, could some oxygen from the metal oxide itself also be involved in the reactions, adding to the oxygen output? That hypothesis has been widely debated but never resolved.
To address that question, the MIT researchers teamed up with Oscar Diaz-Morales and Marc T. Koper at Leiden University in the Netherlands. Together, they have been performing tests that rely on a special form of oxygen that serves as an experimental marker—the isotope oxygen-18 (oxygen-16 is the form that predominates in ordinary water).
To begin their tests, they first immerse a sample metal-oxide catalyst in “heavy” water containing almost entirely oxygen-18 and then perform electrolysis, instigating reactions on the catalyst. During that process, any oxygen atoms that come out of the crystal lattice of the cobalt-oxide catalyst leave behind vacancies that are then filled with new oxygen atoms—in this case, by oxygen-18 atoms from the heavy water.
The researchers next remove the catalyst from the heavy water, place it in ordinary water, and perform the electrolysis process again. They then analyze the oxygen gas that forms using a mass spectrometer that separates different isotopes based on their atomic weight. The results measure how much oxygen-18 is present in the product gases—oxygen that must have been stored in the metal oxide in the first part of the experiment. The researchers performed this test with catalysts made of cobalt and oxygen plus varying proportions of lanthanum and strontium, and the results were markedly different. As shown in the figures below, the versions containing strontium not only showed greater catalytic activity during electrolysis (as measured by current flow) but also yielded different mixes of oxygen products.
Tests with cobalt-oxide catalysts containing lanthanum and strontium
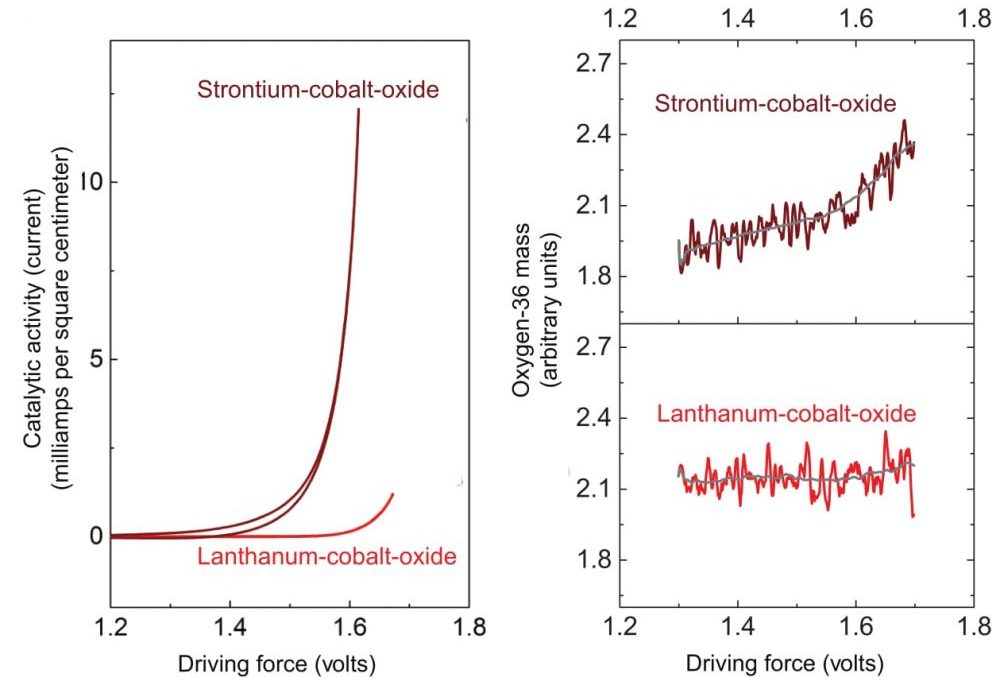
These figures show results of experiments with catalysts of lanthanum-cobalt-oxide and strontium-cobalt-oxide. Left: Here, current flow—an indicator of catalytic activity during electrolysis—is measured as a function of driving force. (Driving force is defined as applied voltage versus a reference electrode, specifically, the reversible hydrogen electrode.) As driving force increases, activity on the strontium-based catalyst far exceeds that on the lanthanum-based catalyst. Right: In the same experiments, concentrations of oxygen-36—formed from oxygen released from the metal oxide lattice—rise significantly with driving force in the strontium tests but remain essentially constant with lanthanum.
In gaseous form, oxygen molecules consist of a pair of oxygen atoms bound together. Analyses of samples collected from experiments with the lanthanum-cobalt-oxide catalyst detected oxygen-32—a combination of two oxygen-16 atoms. The oxygen in the product gases thus came from the water, as dictated by conventional theory.
In contrast, tests with the highly active strontium-cobalt-oxide catalyst yielded products containing a significant amount of oxygen-36, that is, combinations of two oxygen-18 atoms—atoms that can only have come from the crystal lattice of the metal oxide (see the schematic below).
Schematic showing oxygen evolution from the metal oxide lattice
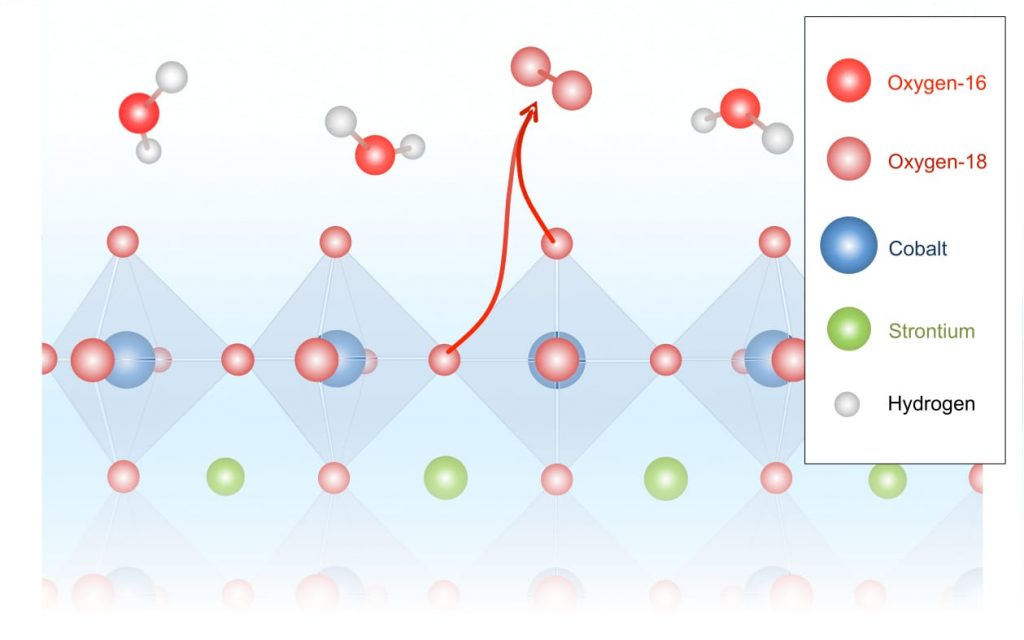
In this schematic, the octahedrons represent the structure of the strontium-cobalt-oxide catalyst, with atoms of interest at their vertices. Water molecules consisting of hydrogen and oxygen-16 atoms come in from above. During the electrochemical reaction, oxygen-18 atoms are released from within the catalyst and pair up, forming the oxygen-36 detected in the experiments.
Those results show conclusively that oxygen in the crystal lattice takes part in the oxygen-evolution reaction. Interestingly, the combined lanthanum-strontium version of the catalyst produced oxygen-34. In that molecular pairing, one oxygen-16 from the water has combined with one oxygen-18 from the metal oxide lattice.
One possibility is that the oxygen-18 is adsorbed and released only on the surface of the catalyst. To check, the researchers calculated how much oxygen-18 could be contained per volume of the strontium-cobalt-oxide catalyst. They then determined how much material would be required to adsorb and release the amount of oxygen-18 they measured in the oxygen gas. Their analysis confirms that the surface layers wouldn’t provide enough storage space. Indeed, the oxygen-18 that’s involved must have come from deep within the strontium-based catalyst.
Tests with the strontium catalyst brought another interesting finding: Changing the acidity of the water caused significant changes in the catalytic activity. With most metal-oxide catalysts—including lanthanum-cobalt-oxide—changing the pH of the water doesn’t affect the reaction rate. But with the highly active strontium-cobalt-oxide catalyst, increasing the pH (that is, making it more alkaline) increased the rate of oxygen evolution.
The figure below summarizes results of these experiments with the catalysts. The impact of changing the pH is almost insignificant with lanthanum-cobalt-oxide, somewhat greater when strontium replaces some of the lanthanum, and quite pronounced in the strontium-cobalt-oxide tests.
Effect of pH on catalytic activity
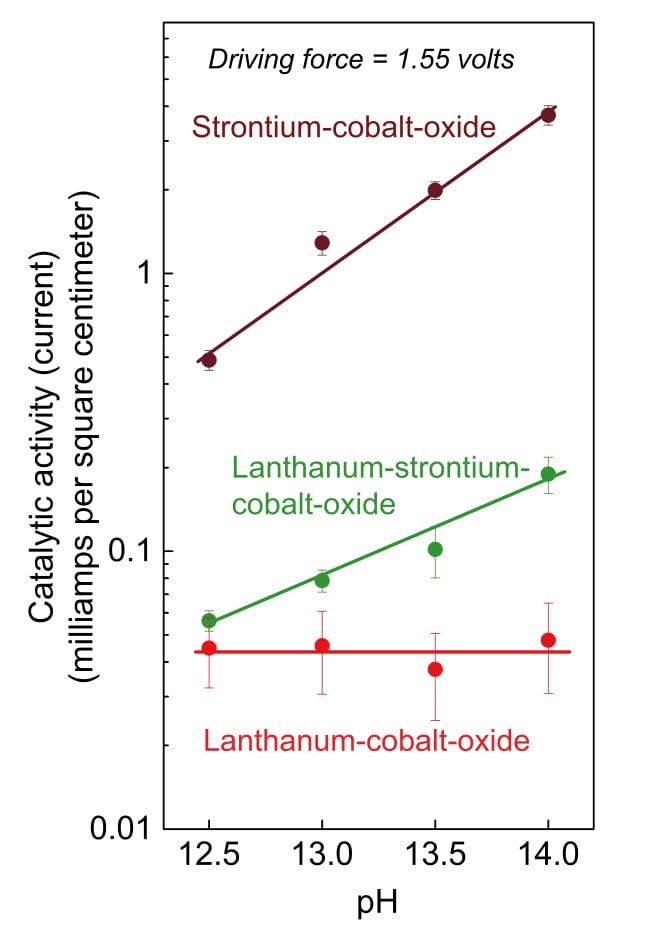
This figure shows how changing the pH of the water affects the catalytic activity of the cobalt-oxide catalysts. While activity on the lanthanum-cobalt-oxide catalyst is unaffected by increasing the pH, adding strontium to the catalyst changes that outcome. In tests with the strontium-cobalt-oxide catalyst, catalytic activity starts out higher and then rises more rapidly as pH increases.
Delving deeper
Clearly, the highly active catalyst doesn’t adhere to the conventional wisdom that all the oxygen for the oxygen-evolution reactions comes from the water and that reactions occur only at metal sites on the surface of the catalyst. What makes the strontium-cobalt-oxide catalyst so different from the lanthanum-cobalt-oxide catalyst?
Based on their theoretical simulations, the researchers conclude that the answer lies in the electronic structure of the two metal oxides: In the strontium version, the metal-oxygen bonds are far more covalent. In any material, there are certain energy levels that electrons can occupy, and electrons at the highest energy level are easiest to remove. In metal oxides with record-high catalytic activity—such as strontium-cobalt-oxide—the highest electron energy levels of the metal and the oxygen match up, and the materials are said to be covalent.
“In our strontium-based catalyst, both the metal and the oxygen are able to contribute electrons at the energy level needed to link pairs of liberated oxygen atoms to form molecules,” says Shao-Horn. “As a result, both the metal and the oxygen in the catalyst are active sites for oxygen-evolution reactions.”
The researchers’ findings suggest a need to revise the theoretical reaction mechanism describing the sequence of chemical changes that occurs during oxygen evolution on a metal-oxide catalyst. Converting water to oxygen on a catalyst requires the transfer of four electrons and four protons. The conventional reaction mechanism says that the electrons and protons have to move at the same time. That is, their transfer must be concerted. “But that mechanism doesn’t allow for participation of oxygen from the crystal lattice or for pH dependence,” says Giordano.
The researchers conclude that when oxygen evolves on a highly active metal-oxide catalyst, there must be non-concerted transfer. Decoupling the transfer of protons and electrons permits involvement of oxygen atoms from the crystal lattice, and it explains why they observed pH-dependent reactivity. “If the mechanism is not concerted—say, a proton goes first followed later by an electron—then the activity can depend on pH,” says Giordano. “When we change the pH, we are changing the concentration of protons in our electrolyte, and that will affect the rate of the reaction—but only if protons and electrons can move independently.”
Design guidance
The new understanding of oxygen-evolution reactions opens up new directions for designing efficient catalysts. For example, researchers can explore other materials—including alternatives to cobalt—to find combinations with higher covalency. “We’ve shown that evolving oxygen from the metal oxide increases catalytic activity,” says Giordano. “So to design particularly active catalysts, we should try to increase the covalency of the metal oxide so as to trigger activation of oxygen in the lattice and enable non-concerted proton-electron transfer.” Under those conditions, influencing reactivity by changing the pH of the water becomes another avenue to explore.
Their findings offer one more practical hint. At the outset of the project, a primary goal was to identify the rate-limiting step in oxygen evolution. The rate of any chemical reaction is determined by the rate of the slowest step in the process. Speeding up that step is thus key to speeding up the overall reaction.
With many metal-oxide catalysts, the speed of water electrolysis is limited by how fast oxygen comes off the catalyst. But in their highly covalent material, the transfer of electrons needed to free the oxygen molecules should be quite easy. “So we speculate that getting the protons off the surface may actually be limiting how quickly the reaction proceeds,” says Giordano. They can’t yet confirm that hypothesis, largely because there’s no consensus on how to theoretically simulate non-concerted reaction steps on these complex materials. “It’s something we’d really like to do,” she says. Their next job is to figure out how.
The research team for this work also included Wesley Hong PhD ’16, former postdoc Yueh-Lin Lee, and Kelsey Stoerzinger PhD ’16. The research was supported by the Skoltech- MIT Center for Electrochemical Energy, the Singapore-MIT Alliance for Research and Technology, the US Department of Energy, and the National Energy Technology Laboratory. Further information can be found in:
A. Grimaud, O. Diaz-Morales, B. Han, W.T. Hong, Y-L. Lee, L. Giordano, K.A. Stoerzinger, M.T.M. Koper, and Y. Shao-Horn. “Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution.” Nature Chemistry, January 2017. DOI: 10.1038/NCHEM.2695.
This article appears in the Spring 2017 issue of Energy Futures.