
In Brief
MIT researchers and their collaborators have demonstrated a novel system using artificial-intelligence techniques to help identify methods of fabricating materials, especially those that look promising in computer simulations. In one test, the system scanned half a million journal articles, recognized those that contained “recipes” for synthesizing various metal oxides, and produced a massive dataset of parameters such as operating temperatures and times. It then scanned the dataset to find patterns that highlight critical parameters. In a test focusing on one compound, the system generated values for certain operating conditions that are realistic but don’t appear anywhere in the literature. Thus, with no guidance, the system leveraged the database to come up with a novel recipe that could be tested in a lab. The researchers are now working to improve the system’s accuracy and to refine its user interfaces so that experts can easily interpret the results, interact with the system, and select directions for further investigation.
During the past decade, much progress has been made in using computer modeling methods to identify new materials with properties optimized for a particular application. Computationally designed materials have been identified for the fields of energy storage, catalysis, thermoelectrics, hydrogen storage, and more.
“Given that we can now identify promising new materials through computer modeling, the bottleneck in materials development has become figuring out how to make them,” says Elsa Olivetti, the Atlantic Richfield Assistant Professor of Energy Studies in the Department of Materials Science and Engineering (DMSE). Synthesizing a new compound suggested by theory can be quite challenging—even in the laboratory. Developing a commercially viable procedure for its large-scale synthesis can take months or even years.
Three years ago, Olivetti and her MIT colleague Gerbrand Ceder, then the R.P. Simmons Professor in Materials Science and Engineering, began discussing whether artificial-intelligence techniques could help identify what Olivetti calls “recipes” for making specific materials. If successful, such an approach could generate recipes for making materials that have been identified through simulation but never before made. And perhaps eventually it could identify more efficient or less costly ways to make known materials.
At about that time, Olivetti learned that Elsevier, a major publisher of scientific journals and books, was opening all of its materials for text- and data-mining activities. She then contacted Andrew McCallum, a professor in the Computer Science Department at the University of Massachusetts at Amherst and a world-renowned expert in natural language processing. Olivetti thus gained access to a wealth of source material and a colleague with the expertise needed to test out her idea.
Joining them in this investigation has been a team including Edward Kim, an MIT graduate student in DMSE; Kevin Huang, a DMSE postdoc; Adam Saunders, another computer scientist at UMass Amherst; Ceder, now a Chancellor’s Professor in the Department of Materials Science and Engineering at the University of California at Berkeley; and Stefanie Jegelka, the X-Consortium Career Development Assistant Professor in MIT’s Department of Electrical Engineering and Computer Science.
A database of recipes
The researchers’ first challenge was to develop a computer-based system that could automatically examine thousands of papers and recognize, extract, and organize information relevant to materials synthesis. Since there are many ways to describe methods within the text, the team couldn’t define restrictive yet comprehensive rules for reliably spotting the relevant words or combinations of words. They therefore developed a natural language processing system that—with training—could learn to perform the task.
During an analysis, the system examines each research paper, identifies paragraphs that include synthesis information, and then classifies words in those paragraphs according to their roles, for example, compound names or operating parameters. By analyzing the collected data, the system can infer general classes of materials, such as those requiring the same synthesis conditions or those with a similar physical structure.
To demonstrate the system’s capabilities, the researchers applied it to half a million journal articles to find those describing the synthesis of various metal oxides. They then prepared the figure below, which shows the heating temperatures used in 12,913 recipes for synthesizing metal oxides. The results appear in four groups based on the number of constituent elements. The binaries combine oxygen with one element, the ternaries with two, and so on. Recipes aimed at making a nanostructured material appear in blue; those targeted at materials made in “bulk” (with no nanostructure) are shown in red.
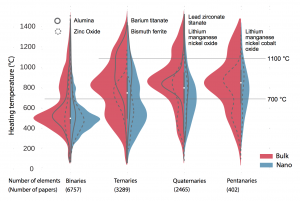
Demonstrating the natural language processing system By analyzing half a million journal articles, the system found almost 13,000 synthesis procedures for making metal oxides. This figure displays the heating temperatures used for making specific compounds, superimposed to form histograms whose width reflects the number of occurrences. Results are divided into four categories based on how many elements are combined with oxygen. Nanostructured target materials appear in blue, “bulk” materials in red. Solid and dashed curves show results for selected compounds in each category. Horizontal rules mark two arbitrary temperatures for ease of comparison. In general, temperatures are lower when fewer compounds are combined and when nanostructured materials are targeted—observations that confirm the system’s ability to pick out and sort relevant data.
Several observations confirm that the retrieved dataset conforms to expectations. For example, the world has published more about alumina made in bulk than alumina made with a nanostructure. “That distribution makes sense because we tend to use alumina as a refractory material, so we’re going to use it more in bulk form,” says Olivetti. The opposite is true for zinc oxide, where nanoarchitectures are required for applications such as sensors and optoelectronics.
The expected correlation between temperature and complexity is also evident. Higher temperatures occur more frequently in cases where multiple metals must mix up and react. And in the nano examples, temperatures are generally kept as low as possible to prevent crystals from growing.
Olivetti notes that this figure is meant only as a demonstration. “The plot was generated from the material our system pulled out of the literature automatically,” she says. “So we used this as a way to say, does what we’re learning make any sense?” And it does.
Mining the database
The researchers’ next task was to mine their database to explore different recipe options and suggest new ones. The natural language processing system maps words or phrases from the vocabulary into real numbers, creating a sort of “recipe space” that includes all the parameters for each recipe—temperature, precursor materials, and so on. But getting guidance from that database is difficult. With just two parameters—say, temperature and pressure—the data can easily be plotted using X and Y axes on a two-dimensional graph. Three parameters can be handled by adding a Z axis, making a three-dimensional space that’s still fairly easy to comprehend. But here there are many, many parameters forming a “high-dimensional” dataset.
While such a dataset may appear unfathomable, it does actually have patterns hidden in it—underlying relationships between synthesis conditions and the materials produced. The key is to find a way to save the information based on those patterns rather than every data point. And that compressed representation must be good enough that it can be expanded to reconstruct the original information without losing any key features.
The best method to perform that task is to use an artificial-intelligence system called a neural network—a technique that’s part of Jegelka’s research in machine learning. A neural network consists of layers of interconnected units, or “nodes,” each of which performs a simple computation and passes the result to the nodes above it. The connections between nodes of different layers are weighted so that each node has a lot, a little, or no influence on the activation of the next node up. Those weights are determined by flowing training data through the network. To train a network for a particular task, the developer selects well-defined datasets where the true output of the analysis is known. The lowest nodes in the network receive numerical inputs, and in an iterative process, the system automatically adjusts the weights until the final output matches desired values. The network is then ready to perform a similar type of analysis on an unknown dataset.
Challenges for neural networks
However, there are challenges with using neural networks to explore materials syntheses. One problem is that there are lots of zeros in the recipe space. Consider just the precursor. For the computer to analyze the dataset, it needs a position (a coordinate) for every possible precursor. But a given recipe will use only a few of the precursors that appear in the literature. As a result, during an analysis of the whole dataset, the computer will encounter lots of zeros—a problem the researchers call sparsity. Such sparse, high-dimensional data points are likewise hard for a human to make sense of.
“Because of our data limitations, we use a relatively simple neural network called an autoencoder that can visualize the data in a way that’s meaningful for a human expert,” says Jegelka. As illustrated in the diagram below, the autoencoder gets the high-dimensional dataset—represented by the blue dots at the left—down to a lower-dimensional dataset—represented by the two dots at the center—that captures the most important aspects of the full dataset. Given an array of input data, the autoencoder learns an encoding model with parameters and weights that can compress the data to two (or a few) dimensions and then a separate decoding model that can take the compressed data as input and reconstruct the original high-dimensional dataset. Since the small number of dimensions at the center restricts the encoding power, the autoencoder is forced to focus on important patterns. Thus, given a full dataset, the autoencoder finds hidden patterns and generates the needed models.
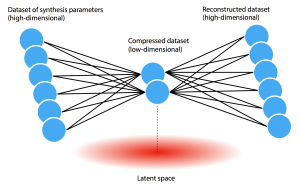
Schematic diagram of the variational autoencoder This diagram shows the architecture of the variational autoencoder. The blue dots at the left represent the high-dimensional dataset of synthesis parameters gathered from the literature. The job of the autoencoder is to find patterns in the data that enable it to compress the full dataset down to two (or a few) dimensions, as shown by the two dots at the center. Given only the compressed data, it must be able to reconstruct the full original dataset. The low-dimensional “latent space” produced during the compression process thus captures key features of the data, generating results that can be displayed on a graph with only X and Y axes.
Based on the center encoding, the model creates a so-called “latent space”—the pink area at the bottom of the figure. In this (two-dimensional) space, each of the original input values is represented as a point that can be plotted on two axes—“latent X” and “latent Y.” “By analyzing a layout of those data points on a graph, we can learn a lot,” says Olivetti.
Jegelka notes one more feature of their neural network: It’s a “variational” autoencoder, which means that the model aims to make the low-dimensional representation of the training data look like a standard distribution—in this case, the familiar bell curve. “As a result, a user can sample a point in the latent space and use just the decoding model to generate a new, realistic synthesis recipe,” says Jegelka.
Augmenting the dataset
Before using their autoencoder, the researchers had to address one more problem: scarcity. While the synthesis dataset is massive, it may actually include only a few recipes for making certain materials, especially new ones. To deal with that issue, the researchers developed a data augmentation methodology that involves training the autoencoder on recipes for producing not only the specified target material but also materials that are similar to it.
In one example, they targeted strontium titanate (SrTiO3), a material used in devices such as capacitors and resistors. Text-mining the literature yielded only 200 temperatures, solvent concentrations, and other descriptors—not enough for a good analysis.
To augment that dataset, they used Word2vec—an algorithm developed at Google—to find materials that appear in similar contexts across journal articles. For example, if recipes for SrTiO3 and another material call for heating to the same temperature, the two materials are linked in the augmented dataset. To further refine the augmented dataset, those related materials are then ranked by how similar they are to SrTiO3 in certain key features of their composition. The higher the similarity, the higher the weight given to those recipes during the SrTiO3 analysis.
As a test, the researchers used the augmented dataset for SrTiO3 plus their variational autoencoder to come up with novel synthesis recipes for making that compound. The autoencoder produced results that mimicked actual recipes well. With no guidance, it defined appropriate categories of operating conditions and generated values for those conditions that look realistic. “So the machine seems to figure out that you need a time and a temperature, and it assigns them in pairs appropriately,” says Olivetti. “We didn’t explicitly tell it that.”
Learning from the latent space
To demonstrate the power of the latent space, the researchers used their overall system to analyze the synthesis of titanium dioxide (TiO2), a material widely used in photocatalysts and electrodes. The latent space produced should reflect the most important features of the full TiO2 dataset. Examining it should therefore show what key parameters drive the synthesis of TiO2 and what factors determine whether the product has a particular crystalline structure. The literature contains many recipes for making the “anatase” and “rutile” form of TiO2 but few for making the “brookite” form, which is of interest as a potential photocatalyst. What causes the brookite structure to form?
To find out, they used their autoencoder to generate the two-dimensional latent space and then plotted the points, producing the figure below. Interestingly, the recipes fall into three general clusters. With further analysis, the researchers determined that the synthesis parameter dominating each cluster was the solvent used: methanol, ethanol, or citric acid. Many of the darkened dots indicating formation of the brookite version fall within the ethanol cluster, but some are elsewhere, suggesting that ethanol may be a preferred but not critical solvent for brookite synthesis.
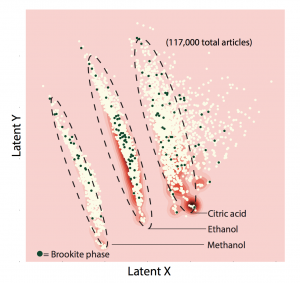
Mapping of two-dimensional latent space from analysis of titanium dioxide synthesis This plot shows results from an analysis of synthesis procedures for making titanium dioxide, with the compressed dataset mapped onto latent X and latent Y axes. The results fall into three distinct clusters. Further analysis showed that the groupings are dominated by the solvent used: methanol, ethanol, or citric acid. Moreover, almost all of the syntheses involving a given solvent fall into the same cluster. For example, 95% of ethanol-using syntheses appear in the ethanol-labeled cluster. The darkened dots indicate syntheses that form a less common crystalline structure called brookite. Many of them fall into the ethanol cluster, indicating that ethanol may be a preferred solvent for making brookite but that other solvents also work.
Olivetti stresses again that the autoencoder identified that pattern on its own. “We didn’t say, give me the solvent that you’re dominated by,” she says. “We just said, plot yourself in this lower-dimensional space.”
Next steps
Thus far, the researchers have been reproducing computationally what’s been observed in theoretical and experimental work. “So we have a new approach, but we haven’t used it to actually make something new based on a method we’ve mined from the recipes,” says Olivetti. “That will be the next thing.”
Olivetti and Jegelka also hope to engage materials-development experts in efforts to further improve their system. Among their goals are finding a way to factor in the knowledge, experience, and intuition of such experts and designing an interface that will allow them to interpret the results and to try out other options. “We aren’t looking to replace conventional theoretical studies and high-throughput experimentation. That’s all still critical,” Olivetti says. “I view our system as another source of information that the experts can leverage in their search for better materials.”
Notes
This research was supported in part by the MIT Energy Initiative Seed Fund Program. Other sponsors include the National Science Foundation, US Office of Naval Research, US Department of Energy’s Basic Energy Sciences Program through the Materials Project, and the Natural Sciences and Engineering Research Council of Canada. Jegelka is supported in part by the Faculty Early Career Development Program of the National Science Foundation. Further information can be found in:
E. Kim, K. Huang, S. Jegelka, and E. Olivetti. “Virtual screening of inorganic materials synthesis parameters with deep learning.” npj Computational Materials, vol. 3, no. 53, 2017. DOI: 10.1038/s41524-017-0055-6.
E. Kim, K. Huang, A. Saunders, A. McCallum, G. Ceder, and E. Olivetti. “Materials synthesis insights from scientific literature via text extraction and machine learning.” Chemistry of Materials, vol. 29, no. 21, pp. 9436-9444, 2017.
This article appears in the Spring 2018 issue of Energy Futures.