
MIT chemists have demonstrated major advances in the design of a device that could one day take carbon dioxide emissions from fossil fuel combustion and—powered by renewable energy—turn them back into high-quality fuels. By examining the individual steps that convert key chemicals inside the device, they’ve identified ways to redesign the catalyst they use so that it selectively encourages the formation of compounds suitable for making fuels. For example, making the catalyst thick and porous significantly increases the production of carbon monoxide, which can be converted to a variety of liquid fuels. Controlling nanoscale features on the catalyst’s surface should further enhance carbon monoxide production. This work provides a compelling demonstration of how fundamental analyses can guide catalyst design—a persistent challenge in many important chemical systems.
Most of the world’s energy needs are met by burning fossil fuels—a process that emits large amounts of carbon dioxide (CO2), a major contributor to climate change. Carbon-free alternatives are now available, notably renewable sources such as solar and wind. But those sources are intermittent, so relying on them at large scale requires a means of storing the excess electricity they generate during peak production for use when the sun and wind aren’t available.
Yogesh Surendranath, the Paul M. Cook Career Development Assistant Professor in the Department of Chemistry, has been working on a way to use that excess electricity to turn CO2 emissions into new carbon-based fuels that could be used when renewable generation is interrupted. The process would provide a means of energy storage for solar and wind systems and would create a closed cycle for fossil fuels. “Right now we combust hydrocarbons, pull more out of the ground, and combust those hydrocarbons, each time making more CO2,” says Surendranath. “If we used renewable electricity to convert captured CO2 emissions back into a fuel, we’d essentially reverse combustion and have a carbon-neutral energy cycle.”
But dealing with CO2 is difficult because it’s chemically inert. One approach to getting it to react is through electrochemistry, that is, using electricity to activate chemical reactions that wouldn’t otherwise happen. Dissolving CO2 plus a salt in water and then activating the mixture with electricity—ideally from a renewable source—would generate hydrogen plus a mixture of carbon-containing molecules.
Unfortunately, that process typically produces far more hydrogen than carbon products, and hydrogen is a less desirable fuel: Gaseous hydrogen stores less energy per unit volume; the infrastructure to support its use doesn’t yet exist; and it doesn’t capture carbon. In addition, CO2 electrolysis produces many different carbon compounds, and separating out the ones that are good starting materials for making fuels is both expensive and energy-intensive.
The MIT team therefore faced a dual challenge. They needed to make the electrolysis system produce more carbon-containing products than hydrogen, and they needed to make it “selective” for the specific carbon products that they wanted.
One determinant of what happens is the catalyst—a material used to speed up chemical reactions involving inert compounds without being consumed in the process. For the past three decades, researchers have been trying to develop better catalysts for CO2 conversion, largely through trial and error and with very limited success. Given all the chemicals present and the many possible reactions among them, unraveling what’s going on empirically is challenging. So Surendranath and his team decided to combine experimental and theoretical studies to develop a detailed understanding of the reactions that take place during CO2 electrolysis.

This electrochemical cell is used for an experiment that both characterizes the gold electrode’s surface and tests its impact on CO2 conversion. The researchers bubble CO2 into an aqueous solution and apply a constant potential to a submerged gold electrode in the left-hand glass cell and then withdraw the gaseous products formed for analysis. Photo: Stuart Darsch
A target compound
As an example, Surendranath, graduate student Anna Wuttig of chemistry, and their international collaborators focused their investigation on carbon monoxide (CO), a good starting material for making liquid fuels, including diesel. Typically, the hydrogen-forming reactions proceed faster than the CO-forming reactions. To find a way to reverse that balance, they needed to understand both reactions in detail.
To that end, they have constructed a lab-scale electrolyzer with an electrode made of gold foil—an element good at both conducting electricity and catalyzing CO2 conversion. They fill their device with a mixture of water and bicarbonate (the salt that carries electrically charged particles through the solution), pressurize the mixture with added CO2, and send in electricity. Products form on the electrode, bubble off, and travel to a gas chromatograph for chemical analysis.
They then use one more tool not generally used in studies of CO2 catalysis: in situ vibrational spectroscopy. While reactions are occurring, they illuminate the catalyst with infrared light, which is absorbed by molecules bound to the surface. The light that is not absorbed is reflected to the detector, and the resulting spectrum is used to infer what molecules are present. This technique allows them to monitor reactions on the catalyst in real time. Indeed, they can “watch” how molecules interact with the surface of the catalyst as CO, hydrogen, and other products are formed.
To generate more CO than hydrogen, the researchers needed to find a way to either speed up the CO-forming reactions or slow down the hydrogen-forming ones. They knew that the hydrogen- and CO-forming reactions both require two types of electrically charged, subatomic particles—electrons, which are delivered by the electrode, and protons, which are delivered by the bicarbonate. But when they figured out all the details of the reactions—the bonds that break and form and rearrange—they found that the electrons and protons play different roles in the two processes, providing a means of controlling the relative rates of the two reactions.
The critical difference
The rate of any chemical reaction is determined by the rate at which the slowest step in the overall process proceeds. In the CO-forming sequence, that “rate-limiting” step involves a CO2 molecule reacting with an electron on the electrode. A proton will be needed, but not until later in the overall CO2 reaction process. In contrast, in the hydrogen-forming reaction, the rate-limiting step requires that an electron react with a proton on the electrode. The two charged particles must therefore be transferred to the electrode surface at the same time.
“That means the rate of the hydrogen reaction on the electrode will be very sensitive to the nearby concentration of protons, whereas the CO-forming reactions won’t be,” notes Surendranath. Since protons are delivered by the bicarbonate, reducing the amount of bicarbonate near the electrode should slow the formation of hydrogen without affecting the conversion of CO2 to CO.
That strategy seemed both simple and promising. But one process inside the electrolyzer could interfere. When bicarbonate mixed with CO2 and water “donates” its protons to a chemical reaction, the CO2 and water could react with each other to make more. If that occurs rapidly near the electrode, it will deplete the CO2 and make more bicarbonate for proton delivery—the opposite of the researchers’ intended outcome.
But that’s not what happens in the vicinity of the electrode inside the electrolyzer. Instead, the bicarbonate concentration remains slightly lower at the electrode surface than in the rest of the mixture. The researchers’ analysis explains why it’s not replenished. The conversion of CO2 to bicarbonate is a notoriously slow reaction—so slow that the CO2 in the area is captured by the electrode and converted to CO long before it can react with water to replenish the bicarbonate concentration.
The porous gold electrode: structure and impacts
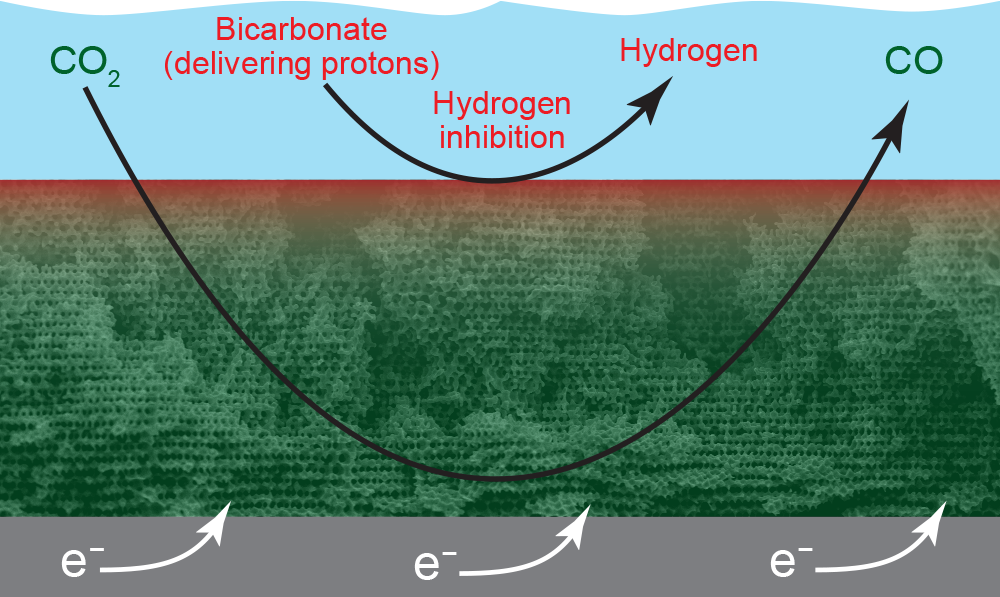
A scanning electron microscope image of the porous gold electrode. Electrons (e–) enter from a metal slab at the bottom; the bulk mixture of CO2, bicarbonate, and water is in the blue region at the top; and dark-colored pores are visible throughout the electrode. CO2 (dissolved in water) easily flows into the electrode, while the proton-bearing bicarbonate is replenished more slowly. As a result, CO-forming reactions occur at reactive sites on the walls of all the pores (indicated in green), while hydrogen-forming reactions occur primarily on the electrode surface (indicated in red).
Porous electrodes
Since the bicarbonate won’t be replenished near the electrode, the researchers just needed to keep fresh bicarbonate from flowing to that area to deliver protons. They’ve now demonstrated a simple way to achieve that goal: They make the electrode thick and porous.
On the thin electrode the researchers originally tested, reaction sites occur only on the surface of the gold electrode. With the thick, porous electrode, active sites are also available on the interior walls of all the pores throughout the volume. But the various chemicals inside the electrolyzer don’t all reach those interior sites with equal ease. As indicated in the image above, CO2 (dissolved in water) flows readily throughout the electrode, so it can react with electrons at all the interior sites. In contrast, bicarbonate diffuses less easily, so it doesn’t reach the interior sites to deliver protons. As a result, CO2 converts to CO throughout the electrode, but hydrogen forms only on the surface, where fresh bicarbonate continues to bathe active sites, delivering protons. The rate of CO formation will thus far exceed the rate of hydrogen formation.
To demonstrate that effect, Surendranath, Wuttig, former MIT postdoc Anthony Shoji Hall, and graduate student Youngmin Yoon of chemistry, a 2016–2017 ExxonMobil–MIT Energy Fellow, fabricated three porous gold electrodes of varying thicknesses and tested them in the electrolyzer. The diagrams below show the measured rates of CO production (left) and hydrogen production (right) at various driving forces with the different electrodes. As expected, the rate of CO production increases with increasing driving force. But at a given driving force, the rate is about the same in all three electrode samples. In contrast, the rate of hydrogen production at a given driving force is not the same in the three samples. The thickest sample consistently shows about a 10-fold decrease in activity relative to the thinnest sample. (The researchers are currently investigating why the curves for hydrogen don’t rise smoothly as driving force increases.)
Effect of porous electrode thickness on CO and hydrogen production
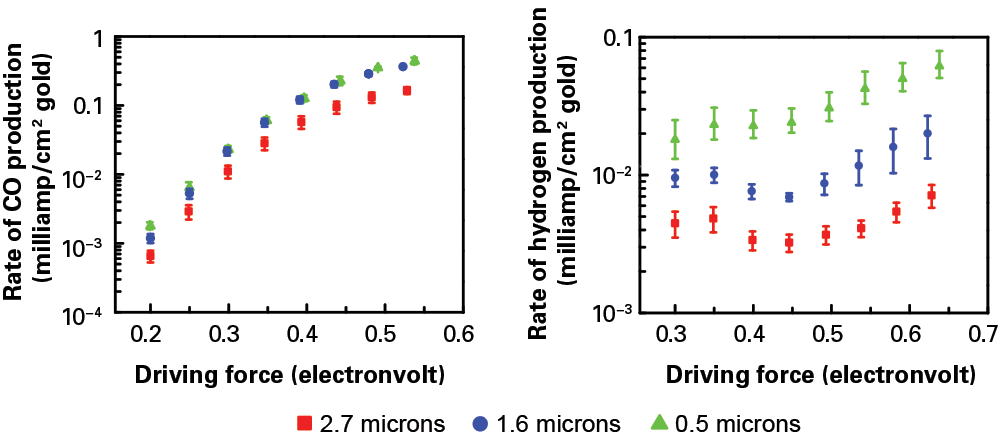
The researchers tested the impact of their porous electrode by preparing three samples of differing thicknesses: 2.7 microns (red squares), 1.6 microns (blue circles), and 0.5 microns (green triangles). Using those electrodes in their electrolyzer, they measured the rate of CO production (left) and hydrogen production (right) per unit area at increasing driving force. At any given driving force, the CO production rate is similar with the three sample electrodes. In contrast, the hydrogen production rate varies significantly, with the thickest electrode exhibiting a 10-fold decrease in hydrogen relative to the thinnest one. The thicker the porous electrode, the more hydrogen formation is suppressed.
“If using a thick, porous electrode leaves the rate of one reaction unchanged but reduces the rate of the other one by an order of magnitude, that dramatically changes the selectivity,” says Surendranath. “Instead of designing a catalyst to speed up a preferred reaction, we’ve made one that slows down the less desirable reaction so that the preferred one can compete.”
Going forward
The researchers are now looking at ways to simultaneously speed up CO production. The in situ spectroscopy analyses showed that about 20% of the surface of their catalyst is covered by stuck CO. Those “spectator” molecules reduce the productivity of the device by covering up sites where other incoming CO2 molecules could react.
While that appears to be bad news, it could actually provide insights into how to design more effective catalysts. The active sites on a catalyst have varied surface features such as terraces and edges, and they bind incoming molecules in different orientations and with varied strength. From the in situ spectroscopy, the researchers can see how the molecules of interest behave on specific sites, so they can see which ones most effectively catalyze the CO2 reactions.
“Ultimately, we may be able to design and manufacture catalysts with surface structures that are optimized for maximum performance,” says Surendranath. “We’re only in the early stages of being able to systematically nanostructure electrochemical catalysts, but that could be a powerful tool for controlling surface sites to build the optimal catalysts for these reactions.”
Recently, they’ve hit upon yet another possible way of improving the performance of their system for CO production. New analyses suggest that varying the diameters and shapes of the pores in their electrode can both reduce the rate of hydrogen formation and increase the rate of CO formation. “Our results to date are a powerful demonstration that elucidating the fundamental behavior of chemicals in a system can lead to important practical insights,” says Surendranath. And he believes there are many more to come.
This research was supported by the Air Force Office of Scientific Research and by the MIT Department of Chemistry. Anna Wuttig was supported by a graduate research fellowship from the National Science Foundation. Further information can be found in:
A.S. Hall, Y. Yoon, A. Wuttig, and Y. Surendranath. “Mesostructure-induced selectivity in CO2 reduction catalysis.” Journal of the American Chemical Society, vol. 137, pp. 14834–4837, 2015.
A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa, and Y. Surendranath. “Inhibited proton transfer enhances Au-catalyzed CO2-to-fuels selectivity.” Proceedings of the National Academy of Sciences, vol. 113, pp. E4585–E4593, 2016.
This article appears in the Autumn 2016 issue of Energy Futures.