
![]() Listen to the bonus interview:
Listen to the bonus interview:
Katie Luu: From MIT, this is the Energy Initiative. I’m Katie Luu. Today we’re talking with Heather Kulik, associate professor of chemical engineering.
Something that really struck me when I was reading the Energy Futures article, the companion piece to this podcast episode, is that you are a chemical engineer who is working with machine learning. Can you tell me how you went down that road?
Heather Kulik: My background is pretty diverse. My undergraduate degree is in chemical engineering. My PhD is actually from MIT in Materials Science and then I did postdoctoral studies in in chemistry so I’d broadly call myself a computational chemist. But that does not mean that we are trained to do machine learning. But it does mean that people in my field that have a strong affinity and our main day-to-day lab instrument is a computer. I came to machine learning from that background that we were already essentially using computers all the time and really the way it started was, like I think a lot of things happen in research these days, I had a student interested in discovering new materials and took a class in machine learning. He asked me what he should do for his homework for his project in the class, and next thing you know it’s what my group is doing for a large portion of full-time.
KL: What are the problems that you have been trying to solve with machine learning in your lab?
HK:: Broadly, my group works on inorganic materials especially transition metal complexes. Those are materials defined by having a transition metal which are elements in the periodic table ranging from iron to cobalt to manganese. These elements that are less common than carbon, nitrogen, and oxygen, maybe, from your undergraduate chemistry class. We’re interested in using these metals and tailoring their properties in order to make better catalysts or materials. A lot of interesting technology comes from being able to precisely design the environment around these metals, to make those metals in turn have interesting optical properties, catalytic properties, and so on.
KL: What kind of uses—end uses—are you aiming for?
HK:: A lot of what my group focuses on is very fundamental in the sense that we’re just trying to develop new ways. If you want to solve a problem in energy or materials or come up with a new technology, where do you start? We start on the very fundamental end of just answering, if the right answer is one in a billion, how are we going to be able to get an idea of what those billion compounds look like, so we can find that one in that space. Where do we need to go? We sit pretty far from the absolute end use of putting this into a new technology but long-term goals are thinking about catalysts that don’t deactivate, that can convert abundant feedstocks like methane to more desirable products like high-value chemicals, transportable liquids like methanol.
Also, in the materials space, there are what was in the Energy Futures article, which is thinking about switchable molecules that can have applications in quantum computing as sensors in functional materials that basically respond to their environment. But thinking about tailoring it on the smallest scale.
KL: Using machine learning, is the whole goal to speed up the discovery of these complexes?
HK:: I would say in the simplest, at the first step, the way we use machine learning is that once you train a machine learning model, it takes us far less time to evaluate what that machine learning model thinks of the next compound. So exactly what you said. In comparison to any kind of experiment in the lab, or what my group had been doing before, which was physics-based modeling. Trying to use laws of physics to solve the properties of the material. The other thing we are increasingly using machine learning for is new ways to think about the molecules themselves.
Once we have a machine learning model and its opinion of how a million molecules should behave, we start to think on a different scale than we normally would, as scientists, when we think about only a handful of molecules. Interpreting what the model thinks, working with the model, it doesn’t just speed up the exploration of the space but it also gives us sort of a map of the space.
KL: What would you say the biggest challenges have been thus far?
HK:: One for sure, that a student of mine and I are working on a little bit, is that machine learning itself is a really exponentially growing area of research. Basically, it went in the past couple of years from no one I know applying it, or very few people applying it, to almost everyone. What that means is there’s some amount of hype cycle that you need to educate your students about but then there’s also the case that an undergraduate education does not necessarily uniformly teach students all the techniques they need to know. My student and I are currently writing a book and have prepared a workshop on machine learning for chemists. I would say that’s one step. I hope to hand that book to my students in the future to simplify some of that.
But I think it’s the wild west right now in terms of how much in a published paper are people reporting the technical details of what they’ve done, you know. Do we all universally agree upon some syntax for explaining what was done? Do we all use the same language? I think there are some challenges there.
KL: When do you hope that your book and workshop will come out?
HK:: The workshop, my students and I, as part of a MISTI grant, put that together last year. The slides are somewhere on the internet. I can probably get a link to you. But the book we’re sending to the publishers for the first draft this month, so hopefully next year.
KL: In the article it talks a lot about neural networks. Could you go into detail about those?
HK:: Sure. Without using too much terminology, neural networks are actually a really old idea and I learned this while writing the book that I just mentioned. Neural networks, artificial neural networks, were something that first captivated people in the 1950s. People were really interested in how the human brain worked and they were interested in building computational models of what they thought a brain was doing. What it was in a neuron was doing. The idea is that you send a signal to a neuron. the neuron responds by turning on, and there are these networks of all of these interconnected neurons and some information about that signal turns it into a final output or result. What’s funny is this died off in the 60s and 70s because the early neural networks people came up with were not complex enough.
What differentiates modern 2010s neural networks from these early 1950s and 1960s neural networks is they have what’s called a “hidden layer”. We started with the 1950s neural network, which has an input and an output. Basically, I send a signal to it and my signal might be anything we think of as a sense we have, but transformed for a computer. Then on the other side we get an output. For instance, input is, “it feels cold out”, output is, “I think I will put on a jacket.” But hidden layers are additional transformations of that signal. Mixing of the inputs, more transformations. Neural network transformations are highly nonlinear so it means that they can make more complex mappings.
But both the theory and the development of how you would build these models lagged for 20 or 30 years until actually in the 1980s and 90s, when people start to develop better algorithms and computers became able to train those models. We’re actually in the second renaissance of neural networks. Which is something I learned recently and why I think it’s cool to mention. If you look if you look back into the scientific literature in the mid-1990s, people were just as excited about neural networks as they are today for exactly the same applications as they are today. The difference is, still then, the kind of things that you could think about applying a neural network to were much smaller in scale. Again, because computing power and complexity weren’t there yet. What I would say is, the reason machine learning and neural networks are ever-present now is largely thanks to advances in computing power. What differentiates a 2019 neural network from a 1960 neural network is how these different neurons are connected and how complex those mappings are.
KL: You gave the example of an input being “it’s cold outside” output being “I’ll put a jacket on.” What does it look like when you add more complexity to it?
HK:: There are, without getting too far into the weeds, there are a lot of ways, for instance, that the most complex models today take a whole series of possible inputs and within the hidden layers, make choices about which ones are the most important. For instance, this becomes very hard to generalize an analogy, but the inputs could be the temperature, how hungry you are, how tired you are, what color your co-worker’s shirt is. The output could be some much more complex processed combination of these inputs that ultimately lead to you deciding to go out to lunch that day. The sum of these models, the most complex ones, essentially learn what is the most important piece of information from those pieces to make the ultimate decision or prediction that they want to make. The hidden layers allow the model to essentially transform the input features much more. In early simple models, even thinking about things much simpler that still have a relationship to neural network like linear regression. A lot of people have fit a straight line through data before, if they’ve completed a math class or done an undergraduate lab. Neural networks are doing that but in a much more complex way. The more pieces there are in the architecture, the more complex that mapping is.
KL: Can you talk more about your process with your team for training and the various rounds of testing you may have done to get to your final outcomes?
HK:: In the piece we actually used a relatively old, what we call representation. That’s what I was calling inputs a few minutes ago. How do we decide to tell a computer what a molecule is? There a lot of ways to do that. That’s actually, as chemists, or as chemical engineers, that is where most of the effort really goes in. In machine learning, there’s the question of, how do you choose to represent the molecule to the computer? How complex is the model you decide to train? How much data do you have? These are all interdependent questions and answers. If you have a lot of data, you can choose a very brute force representation. If you use a very sophisticated model, it can overcome limitations in your representation.
The earliest pieces of that work came from the homework project I mentioned earlier. We started with representations of transition metal complexes based on what I thought I knew as a scientist for about a decade’s worth of training, myself, personal training. I would come up with a list of different possible inputs and then we’d look at how much the model liked them or not. That was through a pretty extended trial-and-error process to train the initial neural network model. We then moved on to a more systematic ways of doing this. We showed that the systematic ways later could reproduce what I thought. I was very proud of my 10 years of experience and what I knew, but we could we could let statistics tell us which of those pieces were right or wrong. I’d say that was one of them more significant pieces of effort in that project.
KL: When you were envisioning this idea and how to develop it over time and get more systemic, did you have some sort of sketching process that you did for this?
HK:: No, I was a relatively new professor. I was just at a workshop where we commented, people who use computers don’t tend to draw things out very much. I was meeting my student every week and literally every week I’d say, “Well, why don’t you try this? This should really work.” Then he’d need to come back and say, “No, that doesn’t really help anything.” When we were moving towards a more systematic approach, we had been reading papers and found that there was a nice representation of molecules people had actually been using since the 80s that that looked like it had a lot of relationship to what we had already been working with. It contained everything we’d been already working with but could extend it. It was a natural fit.
KL: I’d like to go a little bit into your background, if that’s okay with you. You had mentioned earlier that you have materials science training and now you’re in chemical engineering. Can you talk more about your journey and what led you back to MIT?
HK:: I was a postdoc at Stanford University for the most part, working in a computational chemistry group. I never, when I was a student, expected that I would come back here to be a professor. It was very shocking at first. I had to make rules for myself about what I was going to do that was going to be different about being a professor at MIT than a student. For me that rule is you never walk in the tunnels. Tunnels are for students, not for professors.
KL: You mentioned earlier that there is a hype regarding machine learning, artificial intelligence, in the community right now. Do you think people are too excited about its potential or not as excited as they should be?
HK:: There was an article in Chemical Engineering News last year called, “Is machine learning overhyped?” Half of the community answered yes and half of the community answered, basically, it’s not hyped enough. I was one of the people who answered yes. I think I think it’s overhyped. The minute you have to write an article like that the answer is probably yes. But, you know, we found that, I guess, going back even a few years, you know, I’ve seen work that we’ve done in the traditional way get maybe one-tenth of the attention than work we’ve done with machine learning. I think both are equally valuable and so to a certain extent the machine learning work does stand to drown out some traditional work that I think is extremely important.
That being said, going back into where I started as a faculty member. We were really interested at the time in this field called cheminformatics. Cheminformatics was a mature field of the therapeutic drug discovery community basically taking organic structures and knowing based on structure how you could predict a property. Will that be a good drug molecule? Will it get into the bloodstream? We were interested in extending that to transition metal chemistry. Calling it cheminformatics is a lot less trendy these days than machine learning. Cheminformatics itself was trendy for a time. I think for me, I just get a little nervous about, is the community swinging so far one way that it’s going to have a sort of boomerang effect? What I used to tell myself is, every time we apply machine learning in in my group’s research, do we think we’re doing something fundamentally better then we could do through a traditional or more established technique? If I woke up tomorrow and machine learning was a bad word in the community, like saying it in my research meant that people would roll their eyes a little, would I still do what I’m doing? That’s something that I try to ask every time we apply machine learning in our research. To keep us anticipating that at least some applications of machine learning right now are overhyped. There are cases where people are essentially doing linear regression that’s been around forever and calling it machine learning. People are going to get tired of hearing words that are not descriptive enough for the research that’s being done.
KL: Could you provide a few examples of the traditional methods that you’re, you feel are being a little overlooked?
HK:: What’s interesting, or what I would probably describe without too much jargon about machine learning, is that machine learning excels where the relationship you are trying to build or learn is extremely nonlinear. That if you try to write down laws for why A goes to B, you would really struggle. But there are lots of fields. There things like, we know how fast an object will drop if we drop it. There are a lot of things where there are physics based laws where we can predict the outcome deterministically and with a rigorous relationship or rule, will exactly predict the outcome. As a computational chemist, a lot of my community is, what they were doing before they were doing machine learning is developing those rules. Coming up with ways to solve those rules on a computer, developing new rules and insights. That is what I would call the traditional way. Building a relationship that seems rooted in the laws of physics and chemistry that deterministically and analytically gives you why A goes to B. There are fields of science where it’s hard to figure that out, or where the problem is too complex. There are fields of science where it’s easier to figure that out. Discriminating between the two is, I think, important.
KL: What are the key takeaways that you hope folks will have from your machine learning research, in terms of the transition metal compounds?
HK:: I would say for us some of the key things have been, like I mentioned earlier, coming up with a new way to understand structure property relationships. If a complex looks a certain way, what are its properties going to be? One of my, I think, favorite figures to come out of this, which may not have been in the article, is we imagined the properties of a few thousand transition metal complexes. We mapped them onto a picture of a very common transition metal complex known as a porphyrin. A porphyrin is also known heme and it’s responsible for how we breathe. Its binding of oxygen and release of oxygen is how oxygen circulates in our body. It’s a pretty important molecule. With our machine learning models and representations, we’re able to look at which parts of the heme light up to predict different properties of the heme. One is a kind of esoteric property that we’ve spent a lot of time on known as the quantum mechanical ground-state spin. This is important because different spin states of molecules have different macroscopic properties. If our hemes in our body were in a different spin state, then we would not be able to breathe oxygen properly, this type of thing. But then also something else like redox potential which is relevant for energy storage. By building these maps, we’re able to see that the parts of the molecule that light up for redox potential are different from the parts of the molecule that light up for spin. These days we build these maps for everything, for catalysis. We’re able to think, both through the machine learning model and through these property maps, are these two properties that I can design independently to meet an overall design objective, or are they two that are too similar? Once I tune one I’m going to tune the other. That’s really where my group is going in terms of applying these models. We are trying to push machine learning to the point where we can satisfy multiple design objectives in one molecule. In a way that traditional trial-and-error would not work.
KL: Could you define spin for me?
HK:: Spin is a quantum mechanical property of electrons. Electrons are a fundamental particle, the best part of the atom and electrons carry spin. When I say spin of a molecule, what I’m referring to is whether those electrons pair up or whether those electrons like to be unpaired. Traditionally knowing if they want to be paired or not is something that is a really hard property to predict with any with any certainty. Normally the only way to accomplish that is to use methods on a computer that model the electronic structure of these systems. Those calculations take a very long time and so machine learning models that predict spin are advantageous. But here I mean it to say, how many electrons are paired? An electron can have spin up and spin down and if you have two of them and there’s spin up and spin down then they’re paired and there’s no net spin.
KL: It seems like a battery almost? Like spin up is positive and spin down is negative?
HK:: Another analogy to maybe bring it closer to things people are used to thinking about is, basically, all magnets you’ve ever worked with have a permanently aligned net spin of their electrons. If you have a permanent magnet, that means all of the spins always stay and all pointed in one direction with a net spin. If you have something that will magnetize, when you bring it next to, when you bring it next to a magnet but then it loses it, that’s actually all of the electrons aligning in one direction temporarily. Then they’re relaxing their materials properties, meaning that they’re going back and they can be paired up again. If you’ve ever taken like a pin and magnetized it with a with a fridge magnet or something like that. There are interesting materials properties that relate to electronics and energy storage as well, where it may be advantageous to have the spins paired up or it may be advantageous to have the spins aligned. In terms of practical applications, being able to control that spin or switch it and hold it in one direction is really where the community is going for the smallest units of electronics. Where the quantum computing community is headed, for instance, that would be the most obvious practical application beyond fridge magnets.
KL: In terms of energy storage, can you give some examples of where spin would be helpful there?
HK:: I think that will take things down all a little bit of a trickier road. I think the most complex thing to say there is, basically, controlling in which spin channel electrons conduct can be helpful for designing pretty complex electronics. In terms of energy storage, a lot of the most promising redox flow batteries are transition metal complex catholyte and anolytes, with their own net spin and electronic properties. Changing the spin states of those molecules changes their redox capacity. Spin is something that’s always present, it’s just not something that is covered a lot. It’s sort of always in the background when you’re learning some of these concepts early on.
KL: Why do you think that is?
HK:: You’d need to take a year of quantum mechanics to understand, to begin to understand where the spin quantum number comes from. There are things that I think people learn in undergraduate chemistry, such as the Pauli Exclusion Principle, telling them how to pair up electrons or that there are certain rules. We tend to learn things the first time in chemistry and materials as heuristics. Spin is behind a lot of those heuristics but they tend to get written down by, okay, this wants to bond this way or these atoms are going to behave like this. For instance, oxygen, which I mentioned, is one of the most well-known examples. Oxygen has a net spin. The oxygen molecule O2, in its ground state, is a triplet. Which means it has two unpaired electrons. Singlet oxygen would be if we paired up all of the electrons. It can be toxic and damaging to cells and we would not want to breathe singlet oxygen, so spin is there, even in the most fundamental building blocks of life. It’s just not something most people spend a lot of time thinking about.
KL: How does using machine learning affect energy efficiency of computers?
HK:: It’s funny, I think we think about this all the time or it’s always in the background of what we think about as computational researchers. As computing gets more and more powerful, it becomes increasingly possible for me to have a supercomputer running full-time that draws down a lot of energy. What I would say about machine learning that’s interesting, or that we all have to admit as a community, is that we need training data from somewhere. Where my group started is to run those computational chemistry calculations, which are very time-intensive. Then model training, training the machine learning model, can take anywhere from minutes to maybe up to a day, depending on the complexity of the model. Those are both relatively energy-intensive processes. But once trained, the model costs nothing to evaluate. That’s where you start to see the payoff in efficiency. We’re now able to do one-tenth of the calculations we’d normally do.
An interesting example of this, that I hope I can explain clearly, is we actually have built machine learning models designed to take that a step further. We normally use the machine learning model to go out and discover new compounds. But the problem is, the model might take us to a place where the molecule isn’t valid. The molecule might fall apart. Or if a human were to set up that molecule, the human would look at it and say, no that’s not a good idea. I have my PhD. I know you should never simulate that model. In a recent paper for my group, we developed two different ways where we could decide, as a simulation is running, we should… this is a waste of computer time. Stop the simulation. The machine learning model now is not just predicting properties, but it’s predicting, will this simulation be a good use of computer time? We have two models like that. One actually looks at just the molecule, without doing any calculations at all, and says, should you run this calculation, yes or no? These calculations are time -intensive. What they essentially lead to is that we’re able to look at all of the practical molecules that we could characterizes as new leads and spend only a third of the time that we would normally spend on a computer to characterize them. That is that is one way I would say machine learning has led to efficiency directly in a practical way. Though it is a little esoteric.
KL: In the article, it refers to molSimplify. Can you tell us about that?
HK:: molSimplify is my group’s open source code for automating transition metal complex discovery. We put all of our machine learning models in there. It really goes back to that initial goal I mentioned to you, which was bringing cheminformatics to materials.
KL: You said it’s open source, right? Anyone can find it and check it out?
HK:: Yeah. It’s on our website. It’s available via Conda and Github. We’ve actually even used it to teach in my undergraduate/graduate computational chemistry class.
KL: It’s hard to talk about neural networks without mentioning Elon Musk’s Neuralink, which had its first public launch event in San Francisco last month. How similar are these neural networks to the ones that you’re working on?
HK:: My understanding is Neuralink is about bringing the interaction between people and machine learning models even closer. Right now, I think all of us are interacting more and more with machine learning models, but we don’t have electrodes directly implanted in our brains. I think there are, and I’m by no means an expert on it, I think there are interesting questions for us to ask as a society when machine learning models become capable of making decisions or aiding or influencing decisions, and how are we going to interact with them. I think that Neuralink will definitely force those conversations to happen sooner than later. But if those types of things can ultimately help in a lot of neurological diseases, there are a lot interesting questions I think in terms of, can you interpret and help people who are having, you know, things like Parkinson’s. Some of that already exists, where you need a direct feedback. There’s a clear need based on the health condition of need for feedback between what’s happening in the brain and being able to deliver medicine let’s say.
KL: For people who are interested in your area of research, what sorts of reading recommendations would you have for them or recommendations for other resources?
HK:: I think this is an interesting area, in the sense that domain knowledge is always key. I would not underestimate the value of domain knowledge. By which I mean, what is the physics or chemistry or materials problem you’re interested in. But then looking at the connection to machine learning, I think there is, unfortunately, I’m not going to remember the name of the title of the book, there is a book called like “Learn Machine Learning in One Hour” [“Mastering Machine Learning in One Day“] that’s particularly well-written but I don’t think that’s the precise title. I might have to go look it up. But I think there are a lot of great resources on the web, a lot of good online courses. I think I wouldn’t overestimate how expert you need to be to start applying techniques. I think on the low end, there are really useful things to learn about, that have application to even how people are doing traditional science that come out of machine learning or are more central in machine learning. These would be things like, learning to cross-validate. Learning to leave some of your predictions out. When you’re making a prediction, leave some of your test data out and see how well your model actually predicts something. Learning how to do feature selection or regression. Those things are well-covered in lots of basic texts and are a good place to start. You don’t have to start at neural networks, is my long answer. You could just cut to that.
KL: What’s next for you and your group?
HK:: I think the thing that we need to push for the most, is really showing now what we can do with machine learning models once they’re trained. We’ve spent a lot time showing we can train machine learning models. But really the exciting thing is to start to design materials with them in ways that we couldn’t before and for us that means a lot of multi-objective optimization. Which means, I want a molecule that has this perfect property but I also want it to be cheap or I want it to be earth-abundant or I want it to be stable and hold up for a lot of cycles or I want a redox couple that’s also soluble in the electrolyte. Satisfying many different design objectives at once, I think is where the machine learning models are going to shine.
KL: Where can we keep up with your research?
HK:: My group is on Twitter, where we update things a lot. Also, our group’s website. I’d say a lot of the computational chemistry and machine learning communities are on Twitter these days.
KL: Thank you so much for joining us today and talking about your research. It was such a pleasure.
HK:: Thanks for having me.
In brief
MIT researchers have demonstrated a way to rapidly evaluate new transition metal compounds to identify those that can perform specialized functions, for example, as sensors or switches or catalysts for fuel conversion. Using data on known materials, they first trained an artificial neural network to identify good candidates—an approach that’s been used with success in organic chemistry but not previously in transition metal chemistry. They then coupled their trained network with a genetic algorithm that could score each compound, discarding those that are too different from the training data for the neural network to be accurate and too similar to be viewed as new options. Using this procedure, they examined a database of 5,600 possible compounds and identified three dozen promising candidates, some of which were totally unfamiliar to materials scientists. Performing the analysis took just minutes on a desktop computer rather than the years that would have been required using conventional techniques.
In recent years, machine learning has been proving a valuable tool for identifying new materials with properties optimized for specific applications. Working with large, well-defined data sets, computers learn to perform an analytical task to generate a correct answer and then use the same technique on an unknown data set.
While that approach has guided the development of valuable new materials, they’ve primarily been organic compounds, notes Heather Kulik PhD ’09, an assistant professor of chemical engineering. Kulik focuses instead on inorganic compounds, in particular, those based on transition metals, a family of elements (including iron and copper) that have unique and useful properties. In those compounds—known as transition metal complexes—the metal atom occurs at the center with chemically bound arms, or ligands, made of carbon, hydrogen, nitrogen, or oxygen atoms radiating outward (see the drawing below).
Transition metal complexes already play important roles in areas ranging from energy storage to catalysis for manufacturing fine chemicals—for example, for pharmaceuticals. But Kulik thinks that machine learning could further expand their use. Indeed, her group has been working not only to apply machine learning to inorganics—a novel and challenging undertaking—but also to use the technique to explore new territory. “We were interested in understanding how far we could push our models to do discovery—to make predictions on compounds that haven’t been seen before,” says Kulik.
Sensors and computers
For the past four years, Kulik and Jon Paul Janet, a graduate student in chemical engineering, have been focusing on transition metal complexes with “spin”—a quantum mechanical property of electrons. Usually, electrons occur in pairs, one with spin up and the other with spin down, so they cancel each other out, and there’s no net spin. But in a transition metal, electrons can be unpaired, and the resulting net spin is the property that makes inorganic complexes of interest, says Kulik. “Tailoring how unpaired the electrons are gives us a unique knob for tailoring properties.”
A given complex has a preferred spin state. But add some energy—say, from light or heat—and it can flip to the other state. In the process, it can exhibit changes in macroscale properties such as size or color. When the energy needed to cause the flip—called the spin-splitting energy—is near zero, the complex is a good candidate for use as a sensor or perhaps as a fundamental component in a quantum computer.
Chemists know of many metal-ligand combinations with spin-splitting energies near zero, making them potential “spin-crossover” (SCO) complexes for such practical applications. But the full set of possibilities is vast. The spin-splitting energy of a transition metal complex is determined by what ligands are combined with a given metal, and there are almost endless ligands from which to choose. The challenge is to find novel combinations with the desired property to become SCOs—without resorting to millions of trial-and-error tests in a lab.
Translating molecules into numbers
The standard way to analyze the electronic structure of molecules is using a computational modeling method called density functional theory, or DFT. The results of a DFT calculation are fairly accurate— especially for organic systems—but performing a calculation for a single compound can take hours or even days. In contrast, a machine learning tool called an artificial neural network (ANN) can be trained to perform the same analysis and then do it in just seconds. As a result, ANNs are much more practical for looking for possible SCOs in the huge space of feasible complexes.
Because an ANN requires a numerical input to operate, the researchers’ first challenge was to find a way to represent a given transition metal complex as a series of numbers, each describing a selected property. There are rules for defining representations for organic molecules, where the physical structure of a molecule tells a lot about its properties and behavior. But when the researchers followed those rules for transition metal complexes, it didn’t work. “The metal-organic bond is very tricky to get right,” says Kulik. “There are unique properties of the bonding that are more variable. There are many more ways the electrons can choose to form a bond.” So the researchers needed to make up new rules for defining a representation that would be predictive in inorganic chemistry.
Using machine learning, they explored various ways of representing a transition metal complex for analyzing spin-splitting energy. The results were best when the representation gave the most emphasis to the properties of the metal center and the metal-ligand connection and less emphasis to the properties of ligands farther out. Interestingly, their studies showed that representations that gave more equal emphasis overall worked best when the goal was to predict other properties, such as the ligand-metal bond length or the tendency to accept electrons.
Testing the ANN
As a test of their approach, Kulik and Janet—assisted by Lydia Chan, a summer intern from Troy High School in Fullerton, California—defined a set of transition metal complexes based on four transition metals—chromium, manganese, iron, and cobalt—in two oxidation states with 16 ligands (each molecule can have up to two). By combining those building blocks, they created a “search space” of 5,600 complexes—some of them familiar and well-studied, and some of them totally unknown.
In previous work, the researchers had trained an ANN on thousands of compounds that were well-known in transition metal chemistry. To test the trained ANN’s ability to explore a new chemical space to find compounds with the targeted properties, they tried applying it to the pool of 5,600 complexes, 113 of which it had seen in the previous study.
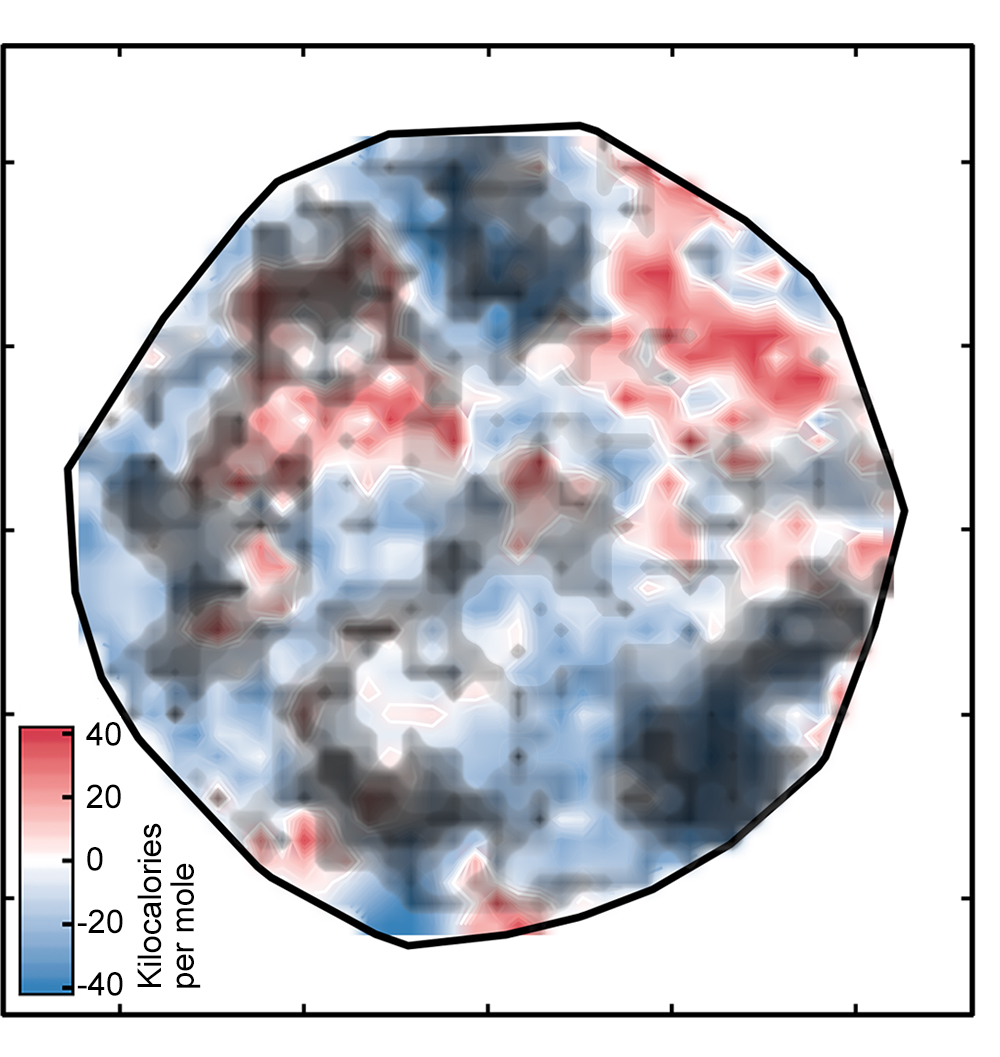
The result was the left plot below, which sorts the complexes onto a surface as determined by the ANN. The white regions indicate complexes with spin-splitting energies within 5 kilo-calories per mole of zero, meaning that they are potentially good SCO candidates (see the key). The red and blue regions represent complexes with spin-splitting energies too large to be useful. The green diamonds that appear in the inset show complexes that have iron centers and similar ligands—in other words, related compounds whose spin-crossover energies should be similar. Their appearance in the same region of the plot is evidence of the good correspondence between the researchers’ representation and key properties of the complex.
But there’s one catch: Not all of the spin-splitting predictions are accurate. If a complex is very different from those on which the network was trained, the ANN analysis may not be reliable—a standard problem when applying machine learning models to discovery in materials science or chemistry, notes Kulik. Using an approach that looked successful in their previous work, the researchers compared the numeric representations for the training and test complexes and ruled out all the test complexes where the difference was too great. Those complexes are covered by the black clouds in the figure on the right below.
Focusing on the best options
Performing the ANN analysis of all 5,600 complexes took just an hour. But in the real world, the number of complexes to be explored could be thousands of times larger—and any promising candidates would require a full DFT calculation. The researchers therefore needed a method of evaluating a big data set to identify any unacceptable candidates even before the ANN analysis. To that end, they developed a genetic algorithm—an approach inspired by natural selection— to score individual complexes and discard those deemed to be unfit.
To prescreen a data set, the genetic algorithm first randomly selects 20 samples from the full set of complexes. It then assigns a “fitness” score to each sample based on three measures. First, is its spin-crossover energy low enough for it to be a good SCO? To find out, the neural network evaluates each of the 20 complexes. Second, is the complex too far away from the training data? If so, the spin-crossover energy from the ANN may be inaccurate. And finally, is the complex too close to the training data? If so, the researchers have already run a DFT calculation on a similar molecule, so the candidate is not of interest in the quest for new options.
Based on its three-part evaluation of the first 20 candidates, the genetic algorithm throws out unfit options and saves the fittest for the next round. To ensure the diversity of the saved compounds, the algorithm calls for some of them to mutate a bit. One complex may be assigned a new, randomly selected ligand, or two promising complexes may swap ligands. After all, if a complex looks good, then something very similar could be even better—and the goal here is to find novel candidates. The genetic algorithm then adds some new, randomly chosen complexes to fill out the second group of 20 and performs its next analysis. By repeating this process a total of 21 times, it produces 21 generations of options. It thus proceeds through the search space, allowing the fittest candidates to survive and reproduce, and the unfit to die out.
Performing the 21-generation analysis on the full 5,600-complex data set required just over 5 minutes on a standard desktop computer, and it yielded 372 leads with a good combination of high diversity and acceptable confidence. The researchers then used DFT to examine 56 complexes randomly chosen from among those leads, and the results confirmed that two-thirds of them could be good SCOs.
While a success rate of two-thirds may not sound great, the researchers make two points. First, their definition of what might make a good SCO was very restrictive: For a complex to survive, its spin-splitting energy had to be extremely small. And second, given a space of 5,600 complexes and nothing to go on, how many DFT analyses would be required to find 37 leads? As Janet notes, “It doesn’t matter how many we evaluated with the neural network because it’s so cheap. It’s the DFT calculations that take time.”
Best of all, using their approach enabled the researchers to find some unconventional SCO candidates that wouldn’t have been thought of based on what’s been studied in the past. “There are rules that people have—heuristics in their heads— for how they would build a spin-crossover complex,” says Kulik. “We showed that you can find unexpected combinations of metals and ligands that aren’t normally studied but can be promising as spin-crossover candidates.”
Sharing the new tools
To support the worldwide search for new materials, the researchers have incorporated the genetic algorithm and ANN into “molSimplify,” the group’s online, open-source software toolkit that anyone can download and use to build and simulate transition metal complexes. To help potential users, the site provides tutorials that demonstrate how to use key features of the open-source software codes. Development of molSimplify began with funding from the MIT Energy Initiative in 2014, and all the students in Kulik’s group have contributed to it since then.
The researchers continue to improve their neural network for investigating potential SCOs and to post updated versions of molSimplify. Meanwhile, others in Kulik’s lab are developing tools that can identify promising compounds for other applications. For example, one important area of focus is catalyst design. Graduate student Aditya Nandy of chemistry is focusing on finding a better catalyst for converting methane gas to an easier-to-handle liquid fuel such as methanol—a particularly challenging problem. “Now we have an outside molecule coming in, and our complex— the catalyst—has to act on that molecule to perform a chemical transformation that takes place in a whole series of steps,” says Nandy. “Machine learning will be super-useful in figuring out the important design parameters for a transition metal complex that will make each step in that process energetically favorable.”
This research was supported by the US Department of the Navy’s Office of Naval Research, the US Department of Energy, the National Science Foundation, and the MIT Energy Initiative Seed Fund Program. Jon Paul Janet was supported in part by an MIT-Singapore University of Technology and Design Graduate Fellowship. Heather Kulik has received a National Science CAREER Award (2019) and an Office of Naval Research Young Investigator Award (2018), among others. Further information can be found in:
J.P. Janet, L. Chan, and H.J. Kulik. “Accelerating chemical discovery with machine learning: Simulated evolution of spin-crossover complexes with an artificial neural network.” The Journal of Physical Chemistry Letters, vol. 9, pp. 1064–1071, 2018. Online: doi.org/10.1021/acs.jpclett.8b00170.
J.P. Janet and H.J. Kulik. “Predicting electronic structure properties of transition metal complexes with neural networks.” Chemical Science, vol. 8, pp. 5137–5157, 2017. Online: doi.org/10.1039/C7SC01247K.
J.P. Janet and H.J. Kulik. “Resolving transition metal chemical space: Feature selection for machine learning and structure-property relationships.” The Journal of Physical Chemistry A, vol. 121, pp. 8939–8954, 2017. Online: doi.org/10.1021/acs.jpca.7b08750.
A. Nandy, C. Duan, J.P. Janet, S. Gugler, and H.J. Kulik. “Strategies and software for machine learning accelerated discovery in transition metal chemistry.” Industrial & Engineering Chemistry Research, vol. 57, pp. 13973–13986, 2018. Online: doi.org/10.1021/acs.iecr.8b04015.
This article appears in the Spring 2019 issue of Energy Futures.
Press inquiries: miteimedia@mit.edu